Revised: Aug 14, 2019
As I discussed in ETFOH, one common theme that cuts across all the autoimmune diseases, and even breast cancer too, is the comorbidity of eczema. At a more fundamental level, and what many others have reported on, is there’s another cross-cutting syndrome, and it’s that of insulin resistance. So much so, that many people feel that insulin resistance is at the root cause of many of the chronic diseases. The very definition of Type II diabetes is insulin resistance and metabolic disease. But, it goes well beyond that. Insulin resistance is hugely implicated in coronary artery disease, heart disease, and surprisingly even the brain diseases such as Alzheimer’s too. You’ve probably heard Alzheimer’s disease even being referred to as Type III diabetes. So, let’s see if we can make some sense out of all of this, and find some plausible common ground and the connections between them.
When I wrote about my theory of obesity causation back in October, I referenced this 2016 report documenting the adipose genesis effect of retinoic acid: Circulating Retinoic Acid Levels and the Development of Metabolic Syndrome.
Source: Yan Liu, Hongen Chen, Di Mu, Jiahua Fan, Jiayi Song, Yuan Zhong, Di Li, Min Xia; Circulating Retinoic Acid Levels and the Development of Metabolic Syndrome, The Journal of Clinical Endocrinology & Metabolism, Volume 101, Issue 4, 1 April 2016, Pages 1686–1692, https://doi.org/10.1210/jc.2015-4038
I was somewhat expecting people to call me out on my use of that study. One of the weird aspects to it was even though the authors documented the adipose genesis effect of retinoic acid; they also concluded that the elevated levels of retinoic acid were beneficial in reducing metabolic syndrome.
Conclusions:
The serum RA level is inversely associated with the development of MetS independently of adiposity and insulin resistance.
So, even though one aspect of that study supported my sub-theory of obesity causation, it directly contradicted my overarching theory that retinoic acid was the real culprit in causing the modern chronic diseases. Of course, there is an absolute mountain of other studies and research proving the incredible toxicity of retinoic acid. To me, at least, something was not adding up here with their contrary findings. But, it is rather easy to see where they go wrong in their analysis.
Here’s the source of the problem stated in the Materials and Methods section of their report:
Determination of RA concentrations
Serum RA concentrations were measured using a commercially available ELISA kit according to the manufacturer’s instructions (catalog number MBS705877; MyBioSource). This assay has high sensitivity and excellent specificity for the detection of human retinoic acid.
Do you see it? They did not actually measure serum RA concentrations at all; rather they measured a biological proxy marker for it. Of course, there’s no such thing as “human retinoic acid” either. So, what are these ELISA kits? They are kits that test for and measure antibodies.
The enzyme-linked immunosorbent assay (ELISA) is a commonly used analytical biochemistry assay, first described by Engvall and Perlmann in 1972.[1] The assay uses a solid-phase enzyme immunoassay (EIA) to detect the presence of a ligand (commonly a protein) in a liquid sample using antibodies directed against the protein to be measured. Performing an ELISA involves at least one antibody with specificity for a particular antigen.
Source: Wikipedia
For example, you could use an ELISA test to detect and measure the antibody proteins to Measles. ELISAs are not usually used to measure and quantify specific compounds such as retinoic acid. Using gas chromatography and mass spectrometry is the more standard technique for identifying and quantifying the molecular makeup of compounds.
Of course, in the human body, most circulating retinoic acid (and retinol too) is bound up in wrapper RBPs. Therefore, what the authors meant to say was: “This assay has high sensitivity and excellent specificity for the detection of the human RBPs for retinoic acid.” So, yes, if the body is building the needed RBPs fast enough, then it will safely wrap up the otherwise highly toxic retinoic acid molecule. When wrapped up in the RBP, the retinoic acid will pose far less of a hazard and people are better protected from it. With this protective mechanism explained we can now understand their observations and paradoxical conclusion.
But, the use of the ELISA test is not some major error on the part of the report authors either. It’s probably a good surrogate test, and it appears to be a rather standard practice. Here’s another report where they’ve done the same, except it’s for retinol RBPs.
Retinol-Binding Protein 4 and Insulin Resistance in Lean, Obese, and Diabetic Subjects
By: Timothy E. Graham, M.D., Qin Yang, M.D., Ph.D., Matthias Blüher, M.D., Ann Hammarstedt, Ph.D., Theodore P. Ciaraldi, Ph.D., Robert R. Henry, M.D., Christopher J. Wason, B.S., Andreas Oberbach, Ph.D., Per-Anders Jansson, M.D., Ph.D., Ulf Smith, M.D., Ph.D., and Barbara B. Kahn, M.D.
N Engl J Med 2006; 354:2552-2563
DOI: 10.1056/NEJMoa054862
Source: https://www.nejm.org/toc/nejm/354/24
Measurement of Serum RBP4
Serum RBP4 was measured by an enzyme-linked immunosorbent assay (ELISA) (ALPCO Diagnostics) in groups 1 and 3 and by quantitative Western blotting with purified human RBP4 standards in group 2. Immunodetection was performed with a polyclonal antibody to human RBP4 (DakoCytomation).
In addition to the ELISA, they are also quantifying their measurements with the Western Blot test. But, like, with the ELISA, the Western Blot is another type of test for measuring antibodies. Next, let’s consider their results:
RESULTS:
Serum RBP4 levels correlated with the magnitude of insulin resistance in subjects with obesity, impaired glucose tolerance, or type 2 diabetes and in nonobese, nondiabetic subjects with a strong family history of type 2 diabetes. Elevated serum RBP4 was associated with components of the metabolic syndrome, including increased body-mass index, waist-to-hip ratio, serum triglyceride levels, and systolic blood pressure and decreased high-density lipoprotein cholesterol levels. Exercise training was associated with a reduction in serum RBP4 levels only in subjects in whom insulin resistance improved. Adipocyte GLUT4 protein and serum RBP4 levels were inversely correlated.
This is an important finding as it is tying together serum RBP4 levels, insulin resistance, diabetes, metabolic syndrome, and more. Except, just for now, ask yourself why are they using antibody tests to measure RBP levels? Doesn’t something about that strike you as being a bit peculiar?
To be clear, these are not procedural anomalies. It appears to be a preferred measurement technique. Here’s another study where they use Western blotting to measure serum retinoic acid levels:
Valproic acid combined with 13-cis retinoic acid and 1,25-dihydroxyvitamin D3 in the treatment of patients with myelodysplastic syndromes
Timo Siitonen, Timo Timonen, Eeva Juvonen, Venla Terävä, Anu Kutila, Tuomo Honkanen, Maija Mikkola, Heikki Hallman, Marjut Kauppila, Pirkko Nyländen, Eira Poikonen, Auvo Rauhala, Marjatta Sinisalo, Merja Suominen, Eeva-Riitta Savolainen, Pirjo Koistinen
Haematologica August 2007 92: 1119-1122; doi:10.3324/haematol.11262
Source: http://www.haematologica.org/content/92/8/1119
Of course, using mass spectrometry to measure RA acids levels is more problematic because the retinoid is entirely encapsulated within the RBP wrapper. Therefore, samples first needs to be dissolved in solvents to breakdown the protein shell and washout the embedded retinoids. From there, mass spectrometry can be employed. So, although the ELISA and Western Blot are measuring a biological proxy marker, they are probably more expedient tests and good enough for the purpose of their studies.
Okay, now we need to really think about this. If these laboratory tests used to measure serum levels of retinoic acid, and even that of retinol, are in actuality antibody tests, isn’t it pretty logical that the RBPs are indeed antibodies?
Except, for the last 50 years now, medical science has claimed that the RBPs are specialized transport proteins. The grand theory is that the liver first scrubs retinol out of serum and then sequesters it away into storage for later delivery. On an on-needed basis, the liver then releases the retinol wrapped up in the RBPs for transport. That’s a nice-sounding theory. But, there’s a huge flaw now showing up in the story. That flaw is because a large percentage of the RBPs are actually being built and released by the adipose tissues, and not just the liver. That’s correct, the origin of much of the RBPs is from the adipocytes.
Conclusions
RBP4 is an adipocyte-secreted molecule that is elevated in the serum before the development of frank diabetes and appears to identify insulin resistance and associated cardiovascular risk factors in subjects with varied clinical presentations. These findings provide a rationale for antidiabetic therapies aimed at lowering serum RBP4 levels.
Okay, it looks like the grand theory of the RBPs being transport proteins is starting to fall apart.
“Until 2005, the sole known function for RBP4 was to mobilize retinol from tissue stores and deliver it to vitamin A-responsive cells where it can be converted to retinoic acid for use in regulating vitamin A dependent transcription and functions. In 2005, Kahn and colleagues reported that circulating RBP4 levels affect glucose clearance, with high RBP4 levels inducing insulin resistance (Yang et al., 2005; Graham et al., 2006). Specifically, Kahn and colleagues proposed that adipocyte-derived RBP4 is a signal that contributes to the pathogenesis of type 2 diabetes, linking obesity with type 2 diabetes, as well as other obesity-related metabolic diseases.”
Very importantly, this then brings into question the entire premise of retinol’s “storage” in the liver. That’s because the thought to be delivery protein for it is being generated by, and secreted from the adipose tissues and not at all exclusively from the liver’s “storage” site!
Next, let’s think a bit about the cell’s response to viral infections. When a cell gets infected with a virus, it can very mistakenly start replicating and cloning more of the same virus. But, very often, it also wraps the newly made viruses up in a protein capsid. Some cells can then eject the entire capsid out of itself. Secondly, in response to viral infections the adaptive immune system will usually start to build antibodies to the virus. The goal and function of the antibody is to attach itself to specific binding sites of the viral protein. In doing so it effectively bulks up any free circulating viruses so that they become too large to pass through the cell membrane. If successful, the viral infection is brought under control. Except, now when a cell is contaminated by a toxin, it is by a non-protein based molecule. There are no protein binding sites (at all) to attach an antibody to it. Therefore, as with some viruses, the cell needs to wrap toxic molecules in a specialized hollow-core antibody (or capsid if you prefer). Unlike the currently recognized Y shaped antibodies, the RBPs are more barrel-shaped, That barrel-shape is needed to engulf the entire antigen. Shown below is a model of the RBP with its embedded retinol ligand.
Structure of human holo plasma RBP4
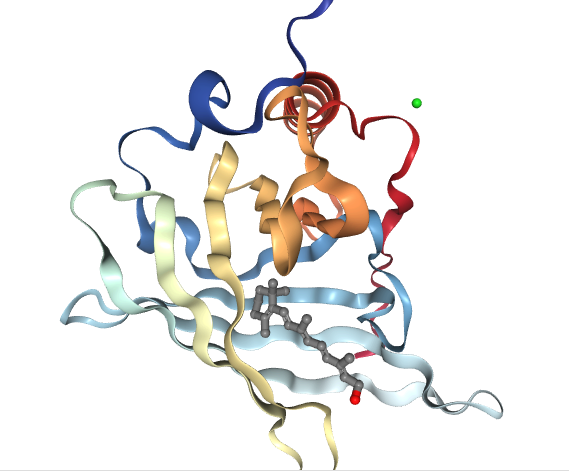
Image created with the PDB ID and associated publication, NGL Viewer (AS Rose et al. (2018) NGL viewer: web-based molecular graphics for large complexes. Bioinformatics doi:10.1093/bioinformatics/bty419), and RCSB PDB.
Source: http://www.rcsb.org/3d-view/5NU7/1
Note the retinol molecule (ball and stick model) nestled inside of the far more complex wrapper (ribbon model) of the RBP. Remember that retinol is too toxic to be in serum alone. Here’s the spacefill model of the RBP showing that no part of the retinol molecule is exposed outside of the RBP itself.
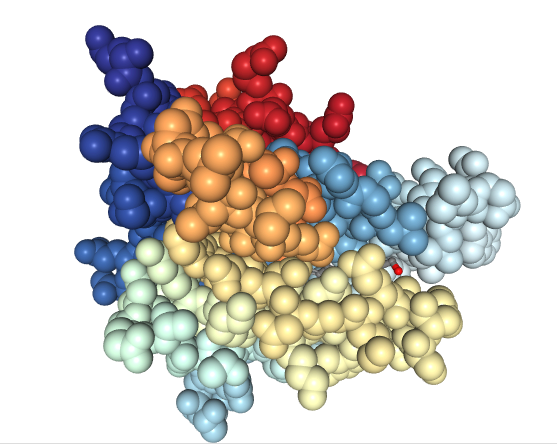
Image created with the PDB ID and associated publication, NGL Viewer (AS Rose et al. (2018) NGL viewer: web-based molecular graphics for large complexes. Bioinformatics doi:10.1093/bioinformatics/bty419), and RCSB PDB.
Therefore, this molecular structure is an astonishing feat of chemical separation and isolation. Not only has the cell’s protein weaving machinery been able to detect and identify a single molecule, but it has also separated it out and wrapped it in a complex protein envelope. Remember too that nature is no fool, and it is not wasteful. This extreme measure of molecular assembly and folding dexterity has to be going on for a very good reason.
Why would the body resort to using such an elaborate and costly transport protein structure? Secondly, if this is truly a transport protein, then how does the receiving cell extract out the retinoid payload when no part of it is exposed to the outside world? And, how exactly does it do it in a toxic-safe manner? Why does the RBP only transport just one molecule at a time? And, once again, why are the adipose tissues generating and secreting the RBPs in the first place? Of course, no one knows the answers to these questions. At best, there are just some unsupported speculations tossed around about it.
“RBP is also bound to a carrier protein, transthyretin. The process by which RBP releases retinol for cellular availability is still unknown and not concisely determined.”
Source: https://en.wikipedia.org/wiki/Retinol-binding_protein
Additionally, if the RBPs are “transport” proteins, that are supposed to be so highly-regulated, then why do they accumulate with age?
The real clincher question is: if the liver is building RBPs to deliver retinol to the other tissues, then why are those receiving tissues then themselves wrapping up retinol in their own internally built RBPs and ejecting it? The current theory of the RBPs being “transport and delivery” proteins makes no logical sense.
Isn’t it just so much more logical that the RBPs are hollow-core antibodies? Isn’t it just more logical that as the body gets exposed to more of a toxic load outside of liver storage, the body builds more antibodies to combat and protect us from that now adipose accumulating toxin? Here’s another study indirectly indicating that’s the case:
Summary
Cellular retinol-, retinaldehyde- and retinoic acid-binding proteins were localized in rat retina during pre- and postnatal development by indirect immunofluorescence.
Source: Immunolocalization of cellular retinol-, retinaldehyde- and retinoic acid-binding proteins in rat retina during pre- and postnatal development
De Leeuw, A.M., Gaur, V.P., Saari, J.C. et al. J Neurocytol (1990) 19: 253. https://doi.org/10.1007/BF01217303
What is immunofluorescence?
Immunofluorescence is a technique used for light microscopy with a fluorescence microscope and is used primarily on microbiological samples. This technique uses the specificity of antibodies to their antigen to target fluorescent dyes to specific biomolecule targets within a cell, and therefore allows visualization of the distribution of the target molecule through the sample.
Clearly, the RBPs are very antibody-like. But, since we now know they are not “transport” proteins, is it not highly likely they are indeed antibodies? They have just not yet been recognized as such. The next logical question is: if the RBPs are, in actuality, antibodies, then what does that tell us about retinol? It tells us that it is a toxin!
The famous amyloid plaques of Alzheimer’s disease
One of the key neuropathological hallmarks of Alzheimer’s disease is the accumulation of β-amyloid plaques in the brain. No one knows why they develop or what to do about them. What’s assumed is that they are just defective ribbon structured proteins that exhibit some weird and unexplained misfolding. But, doesn’t that sound a lot like the folded ribbon proteins of the RBPs? Here’s a description of the RBPs
Lipocalins
Members of the lipocalin family, including the retinoid-binding proteins RBP, epididymal retinoic acid-binding protein and β lactoglobulin, share a very low sequence identity but display a highly conserved overall fold. They are composed of an eight stranded antiparallel β-sheet that is folded over itself to form a hydrogen-bonded β-barrel, which constitutes the ligand binding pocket (Figure 2). The N-termini of these proteins are folded around the back of the barrel, `capping’ that side of the pocket.
Next, we need to know and deeply appreciate that it’s not just the liver and adipose tissues that are excreting the RBPs. It’s also happening in the kidneys, lungs, heart, skeletal muscle, spleen, eyes, and testes too.
Functions of RBP
Retinol is secreted from its storage pools and circulates in blood bound to RBP. The main storage site for vitamin A and, correspondingly, the main site of synthesis of RBP, is the liver, although other tissues (including adipose tissue, kidney, lung, heart, skeletal muscle, spleen, eye and testis) express this protein.
Source: Retinoid-binding proteins : mediators of retinoid action
Biochem. J. (2000) 348, 481±495 (Printed in Great Britain) 481
REVIEW ARTICLE
Noa NOY1
Division of Nutritional Sciences, Cornell University, Ithaca, NY 14853, U.S.A
And then there’s this statement:
The mechanism by which retinol initiates secretion of RBP from cells is unknown, but appears to be conserved in yeast ectopically expressing RBP.
Source: As above
I think it is quite obvious that the cells of multiple tissue types are synthesizing their own RBPs as a protective measure in an attempt to rid themselves of a highly toxic molecule. Do you see where I’m going here? Yes, if the cells of the liver, adipose tissues, kidney, lung, heart, skeletal muscle, spleen, eye, and testis are all able to secrete RBPs, with the retinoid wrapped up inside of it, why shouldn’t we think that the nerve cells of the brain are not going to do the same? Just like the fact that the RBPs are unrecognized as antibodies, couldn’t the famous amyloid plaques of Alzheimer’s disease be unrecognized as RBPs generated by the brain too? Or maybe, since Alzheimer’s is such a new phenomenon in human existence, the amyloid plaques are an ongoing attempted synthesis of RBPs? Thinking that the cells of the nervous system can generate amyloids is not just speculation. As mentioned above, the RBPs are often bound in serum to transthyretin.
Transthyretin
Transthyretin (formerly known as prealbumin) is a plasma protein involved in the transport of thyroxine and retinol and is secreted in great amounts by the choroid plexus epithelium.
Source: Modern Surgical Pathology (Second Edition), 2009
Knowing that transthyretin is typically bound up with the RBPs it’s no surprise that so many people with diabetes, autoimmune diseases, and even dementia have the comorbidity of abnormal thyroid functions.
There are some rather remarkable similarities between the so-called defectively folded proteins of the amyloid plaques and that of the folded over proteins of RBPs. For example, here’s the CRYSTAL STRUCTURE OF THE PROTEASE INHIBITOR DOMAIN OF ALZHEIMER’S AMYLOID BETA-PROTEIN PRECURSOR
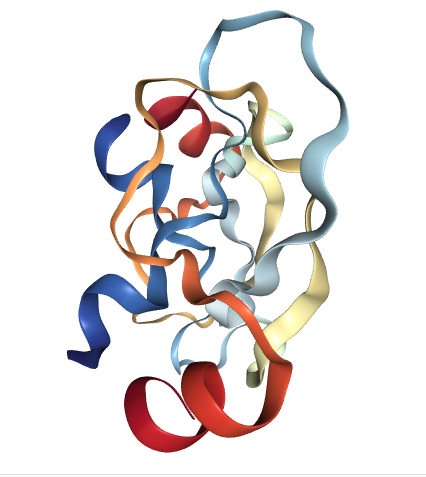
Image created with the PDB ID and associated publication, NGL Viewer (AS Rose et al. (2018) NGL viewer: web-based molecular graphics for large complexes. Bioinformatics doi:10.1093/bioinformatics/bty419), and RCSB PDB.
Source: http://www.rcsb.org/3d-view/1AAP/1
As with the RBPs, the Alzheimer’s disease defining proteins adopt a β‐sheet structure and sometimes form into amyloid plaques. Most importantly, when you consider that there has been an 800 times increase in the rate of Alzheimer’s disease since the early 1970s, it is completely, absolutely, undeniably, positively crystal clear that the disease is a poisoning. It’s then easy to appreciate that the cells of the brain are responding to that poisoning in very similar ways as do say the cells of the kidney, lungs, etc. However, once trapped inside the brain, there is no viable disposal pathway for these unusual proteins. The brain just slowly accumulates and clogs up with the amyloid plaques. Not only that, but all the energy and resources wasted in building the amyloid plaques has been taken away from the brain’s normal tissue maintenance and repair processes.
Very tragically, billions of taxpayer-funded dollars have now been spent by researchers going on a wild goose chase of blaming genetics for this disease. Of course, it is a colossal waste of money and time because it is completely obvious Alzheimer’s disease is not caused by genetics. Not only does it take thousands of years for a genetic shift to occur in the human population, the country with the highest genetic diversity on the planet now has the highest rate of Alzheimer’s disease in the world. And who cares about some supposed “genetic predisposition” to the disease because there’s absolutely nothing people are going to be able to do regarding their genetic makeup. So, investigating genetics is pretty much a meaningless distraction from the real culprit of the disease being environmentally caused. If people were not being slowly poisoned by some environmental factors, then their “genetic predisposition” would never be a problem.
Of course, it’s not just the brain that’s clogging up with amyloid plaques, it’s the heart and other organs too. Not at all surprisingly, that also directly correlates to the serum levels of the RBPs!
Identification of Transthyretin Cardiac Amyloidosis Using Serum Retinol-Binding Protein 4 and a Clinical Prediction Model
Findings In this case-control study that included 34 patients with transthyretin cardiac amyloidosis and 77 control participants, retinal-binding protein 4 was significantly associated with the disease independently of tested confounders. A prediction model composed of retinal-binding protein 4, transthyretin, and echocardiographic and electrocardiographic characteristics had excellent discrimination for transthyretin cardiac amyloidosis (deposition of amyloid).
Source: AMA Cardiol. 2017;2(3):305-313. doi:10.1001/jamacardio.2016.5864
Conversely, the vascularization of the brain in AD patients often shows the same type of damage as that presented in coronary artery and heart disease. The condition is called “vascular dementia.” Therefore, it’s very probable that the same root cause force behind that vascular damage is driving both diseases. Equally important is to know that almost no one dies directly from Alzheimer’s, rather they die with Alzheimer’s. Meaning, the real cause of death is one of their other conditions, such as kidney disease, heart disease, stroke, choking, pneumonia, etc.
Additionally, It’s well documented that persons with Type II diabetes are at a bigger risk (~200%) for developing Alzheimer’s disease. It is even claimed that the single biggest risk factor for developing Alzheimer’s disease is having Type II diabetes. Therefore, many people have made the erroneous assumption that the diabetes is somehow causing Alzheimer’s disease. But, that’s not quite true. Diabetes is not “causing” the Alzheimer’s. Rather, both diseases are simply joined at the hip, metaphorically speaking. And in both diseases we have the common themes of insulin resistance and the development of amyloid plaques. Obviously, diabetes is not “causing” the Alzheimer’s and Alzheimer’s is not “causing” the diabetes. Once again, something else is at the root cause of both diseases. As we’ve seen, elevated serum levels of the RBPs are a significant marker in both diseases too. It is also true for the development of amyloid plaques of the heart. So much so, that it is pretty much an undeniable fact that the RBPs are somehow involved. But, there is an even more direct connection between these two major diseases. As with heart disease, it’s the accumulation of misfolded proteins.
Protein misfolding and aggregation in Alzheimer’s disease and type 2 diabetes mellitus.
AD is characterized by the accumulation of amyloid-β (Aβ) in the brain, while T2DM is characterized by the deposition of islet amyloid polypeptide (IAPP, also known as amylin) within beta-cells of the pancreas.
Source: https://www.ncbi.nlm.nih.gov/pubmed/25230234
And then a bit later, they state:
It is becoming increasingly believed that islet amyloidosis is the progeny of many diseases, including T2DM. The production of islet amyloid polypeptide (IAPP) oligomers that results in amyloid deposition is considered as a major contributor in pathogenesis and progression of T2DM [16]. This has been found present in 96% of T2DM patients and is recognised as a hallmark for diagnosis of this disease.
And then:
LINKAGE BETWEEN AD AND T2DM
AD and T2DM are both prevalent in the aged population. Whereas the cerebral accumulation of Aβ is a major pathological hallmark of AD [168], the deposition of a very different polypeptide, amylin, that likewise succumbs to β-sheet formation and self-aggregation occurs within the pancreas, especially in β-cells, in T2DM.
So now, just what do you suppose is responsible for causing the messed-up structure of the proteins generated by the pancreatic and other tissue stem cells? How about we seriously consider it to be a toxic molecule that has now been proven to cause about 500 different gene “expressions” in stem cells. Except, to most of us here it’s now clear these are, in actuality, sites of gene “damage” and not that of gene “expressions” at all. That gene damage then messes up the cell’s protein weaving machinery. Naturally, the follow-on consequence is going to be the defective structures of the manufactured proteins.
Somewhat like with the wild goose chase of genetics, there’s been a vast amount of research in trying to correct the misfolding proteins using pharmaceutical drugs. Of course, that effort has been futile. Additionally, most research is narrowly focused right down into trying to decipher what’s wrong with the protein structure, Oddy, it appears most people are only asking why the protein is misfolding, as if something has gone wrong with it post manufacturing. They they are not considering that it has been expressly manufactured that way due to RNA/DNA damage.
The RBPs tied-together with Insulin Resistance
GLUT4 – is the glucose transport protein used by cells to take-up insulin from serum. Obviously then if the GLUT4 proteins are not of sufficient quantity or quality, it then will cause the condition of Insulin Resistance. It’s not hard to imagine that if the cell’s energy and resources are tied up generating RBPs, that could be impairing their production and or functioning of the GLUT4 proteins. This scenario is indeed being demonstrated in clinical research. For example:.
Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes.
Yang Q1, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, Kotani K, Quadro L, Kahn BB.
We show that serum RBP4 levels are elevated in insulin-resistant mice and humans with obesity and type 2 diabetes.
Source: Nature. 2005 Jul 21;436(7049):356-62.
This now brings us full circle to where I hope it shows how it’s all interconnected. The information presented here not only links the RBPs to Type II diabetes and the amyloid-β plaques; it seriously implicates it in causing coronary artery, heart disease, and of course Alzheimer’s too. Except, we need to be very careful here and not try to blame the RBPs. Rather it’s the overload of retinol that is the very root cause. It’s the overload of retinol that has forced production of the RBPs in the first place. There’s one more connection with insulin resistance that I’d like to discuss, and that is Mitochondrial Dysfunction. There are hundreds of research papers documenting that Mitochondrial Dysfunction and Diabetes are very closely coupled. But, as always, there’s more to the story, and that is the key role of the retinoids directly damaging the mitochondria.
Vitamin A and Retinoids as Mitochondrial Toxicants
Overall, such findings indicate a potential ability of vitamin A and its derivatives to negatively interact with biological membranes, an event that may lead to organelle stress, as, for instance, mitochondrial dysfunction, and to cell apoptosis or necrosis.
Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4452429/
And:
‘Vitamin A and its derivatives, the retinoids, disrupt mitochondrial function by a mechanism that is not completely understood. However, it accounts with impaired electron flux between the complexes of the METC, increased ROS production, and induction of oxidative and nitrosative stress to mitochondrial membranes. Additionally, vitamin A and retinoids alter the mitochondrial structure by causing swelling of the organelle. More investigations are needed to elucidate how vitamin A and retinoids affect mitochondria and whether there is a causative link between such event and the clinical manifestations observed both experimentally and in humans.’
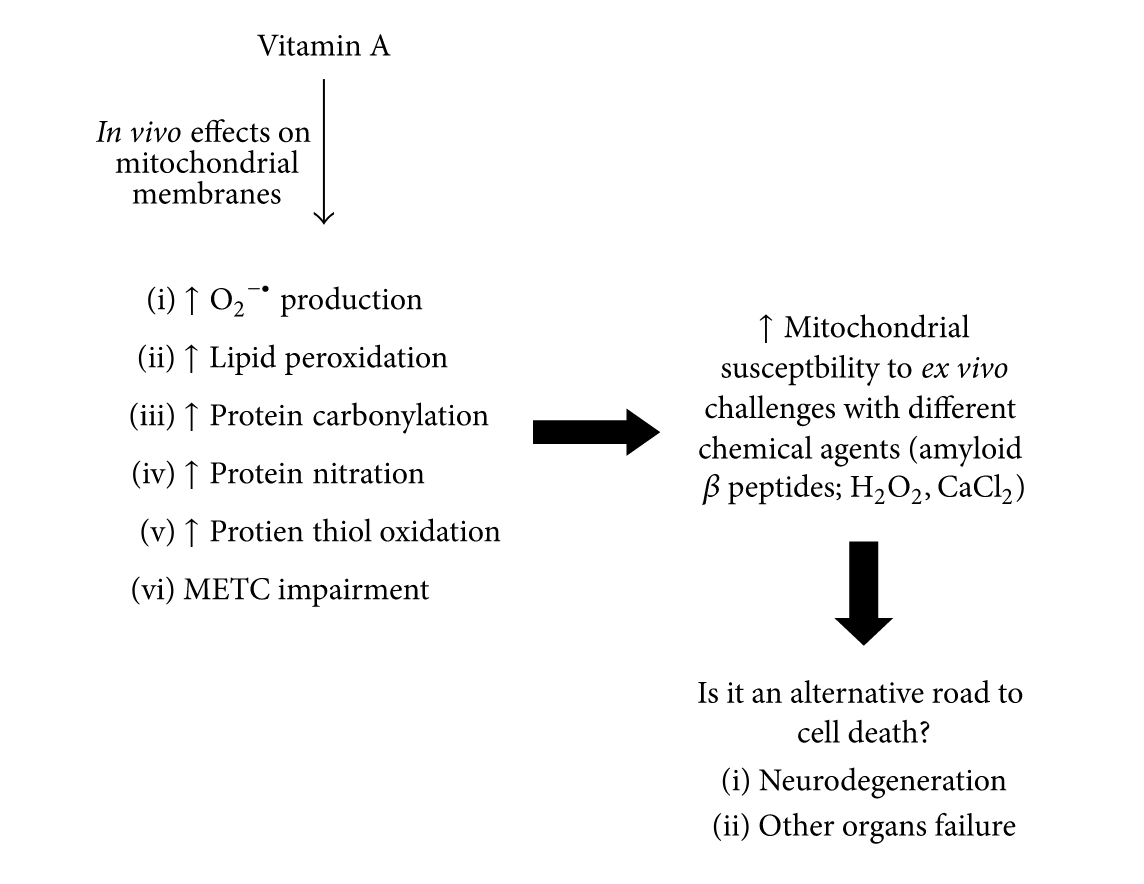
Source: as above
Predictably, the thinking from some pharma funded researchers is the same. It’s viewed as an opportunity to try to develop a new drug that will block or intervene in the synthesis of the RBPs. Of course, it always comes down to needing a “drug” to “block” something. Using a non-drug based therapy is completely unthinkable to these folks. But, since we know with 100% certainty that Alzheimer’s is the result of a long term poisoning, the only “drug” that could even possibly work is one that acts as an antidote to whatever that poisoning is. Until then, almost all other drugs are just going to be useless, and most will cause even more harm. Now with the known connections between the elevated RBPs and the development of amyloid plaques, and having a better understanding of RBPs likely being an antibody response to a toxin, isn’t it very likely retinol is the responsible toxin?
Maybe it’s just me, but how about we just consume a lot less of the retinoids and let the body heal itself?

Are the RBPs antibodies? Please have a think about it and comment as you see fit.
Thanks
Pingback: The Dance of Zinc (Deficiency) & Copper (Toxicity) in Your Body – Nutrition Detective – Research Forum